Multimodal, Multisubject data fusion¶
fMRI Preprocessing and Statistics¶
We will keep description of the preprocessing and analysis of the fMRI
data to a minimum, given that fMRI analysis is covered in several other
chapters. We start with preprocessing Subject 15’s data. For the fMRI
experiment, there were 9 runs (sessions), rather than the 6 in the M/EEG
experiment. This is because the SOA was increased in the fMRI
experiment, by virtue of jittering, which is necessary to estimate the
BOLD response versus interstimulus baseline (e.g., to compare with the
evoked EEG/MEG response versus prestimulus baseline). If you already
launched SPM to analyse the M/EEG data from the previous sections,
select “FMRI” instead of “EEG” in the pulldown menu of the main SPM
window, otherwise start SPM by typing spm fmri. Preprocessing involves
the following modules, which can be found under the menu “SPM –
Spatial”:
Realignment of EPI (fMRI) data¶
The first step is to coregister the 9 runs of 208 images to one another
(i.e., correcting for movement) using a rigid-body transform. Select
“Realign: Estimate & Reslice”, and on the “Data” item, add nine new
“Sessions”. Then, for the first session, select the 208 f*.nii images
in the BOLD/Run_01 directory of Subject 15 (using right-click, “Select
All”), and then repeat for the remaining eight sessions. On the “Reslice
Options” item, change the default value of “All Images + Mean Image” to
“Mean Image Only”. This is because we do not re-slice the EPI data: the
coregistration parameters will be stored in the header information of
the NIfTI file, and can be combined with the normalisation parameters
when re-slicing the normalised images below (re-slicing can introduce
interpolation artifacts so generally best to reduce number of
re-slicings). However, we do need a re-sliced mean image, which we can
use for coregistration with the T1 below. Thus the contents of the
f*.nii files will change (as header updated), but no new (rf*.nii)
files will be output, except for the meanf*.nii file.
Normalisation/Segmentation of T1 images¶
We will use SPM12’s unified segmentation to estimate normalisation
parameters to MNI space. Select “Normalise: Estimate” item, add a new
“Subject” in “Data” and select the mprage.nii image in the “SMRI”
directory of Subject 15 for “Image to Align”. The output of this step
will be a file y_mprage.nii containing the estimated deformation field
that warps data from subject to MNI space.
Coregistration of mean EPI (fMRI) to T1 (sMRI)¶
Because we have determined the normalisation parameters from the
subject’s native space to MNI space via the unified segmentation of
their T1 image, we need to coregister all the EPI images to that T1
image, so that the normalisation warps can be later applied. Select
“Coregister - Estimate” item, and for the “Reference Image”, select the
same mprage.nii image in the SMRI directory. For the”Source Image”,
select “Dependency” and then use “Realign: Estimate & Reslice: Mean
Image”. For “Other Images”, select “Dependency” and then use the Ctrl
key to select all 9 sessions from the “Realign” stage.
Application of Normalisation parameters to EPI data¶
We can now apply the normalisation parameters (warps) to each of the EPI
volumes, to produce new, re-sliced wf images. Select “Normalise –
Write” item, and for the “Deformation Field”, use select the “Normalise:
Estimate: Deformation” dependency. For the “Images to Write”, select the
“Coregister: Estimate: Coregistered Images”. You can also change the
default voxel size from [2 2 2] to [3 3 3] if you want to save
diskspace, since the original data are [3 3 3.9] (for Subject 15), so
interpolation does not really gain new information.
Smoothing¶
Finally, we smooth the normalised images by an 8mm isotropic Gaussian
kernel to produce swf* images. So select the “Smooth” item, and select
the input to depend on the output of the prior “Normalisation: Write”
stage.
Save batch and review¶
You can save this batch file (it should look like the
batch_preproc_fmri_job.m file in the SPM12batch FTP directory), and
then run it. You can inspect the realignment parameters, normalisations,
etc. as described in other chapters. Make a new directory called Stats
in the Sub15/BOLD directory.
Creating a 1st-level (fMRI) GLM¶
Select the “fMRI model specification” option from the “SPM – Stats”
menu. Select the new Stats directory you created as the output
directory. Set the “Units for design” to “seconds” (since our onsets
files are in units of seconds) and the “interscan interval” (TR) to 2.
Then under the “Data & Design” option, create a new Session, and then
select all the swf*.nii images in the Run_01 directory as the
“Scans”. Then under the “Multiple conditions” option, press “Specify”
and select the file run_01_spmdef.mat that has been provided in the
Trials sub-directory. This is a MATLAB file that contains the onsets,
durations and names of every trial in Run1 (for this subject). Then
under the “Multiple regressors” option, press “Specify” and select the
file matching rp*.txt in the Run_01 directory. This is a text file
that contains the 6 movement parameters for each scan, which was created
during “Realignment” above, and we will add to the GLM to capture
residual motion-related artifacts in the data.
For the basis functions, keep “Canonical HRF”, but change the “model derivatives” from “no” to “time and dispersion derivatives” (see earlier Chapter manuals). Then keep the remaining options as their defaults.
You then need to replicate this for the remaining 8 sessions, updating all three fields each time: i.e., the scans, conditions and (movement) regressors. It is at this point, that you might want to switch to scripting, which is much less effort – see e.g. this:
swd = '.../Sub15/BOLD'; % folder containing Subject 15's fMRI data
clear matlabbatch
matlabbatch{1}.spm.stats.fmri_spec.dir = {fullfile(swd,'Stats')};
matlabbatch{1}.spm.stats.fmri_spec.timing.units = 'secs';
matlabbatch{1}.spm.stats.fmri_spec.timing.RT = 2;
for r=1:9
matlabbatch{1}.spm.stats.fmri_spec.sess(r).scans = ...
cellstr(spm_select('FPList',fullfile(swd,sprintf('Run_%02d',r)),'^swf.*\.nii'));
matlabbatch{1}.spm.stats.fmri_spec.sess(r).multi = ...
cellstr(fullfile(swd,'Trials',sprintf('run_%02d_spmdef.mat',r)));
matlabbatch{1}.spm.stats.fmri_spec.sess(r).multi_reg = ...
cellstr(spm_select('FPList',fullfile(swd,sprintf('Run_%02d',r)),'^rp_.*\.txt'));
end
matlabbatch{1}.spm.stats.fmri_spec.bases.hrf.derivs = [1 1];
spm_jobman('interactive',matlabbatch);
Model Estimation¶
Add a module for “Model estimation” from the “SPM – Stats” menu and
define the SPM.mat file name as being dependent on the results of the
fMRI model specification output. For “write residuals”, keep “No” and
stick to “Classical” estimation.
Setting up contrasts¶
To create some contrasts, select “Contrast Manager” from the “SPM –
Stats” menu. Define the SPM.mat file name as dependent on the model
estimation. The first contrast will be a generic one that tests whether
significant variance is captured by the 3 canonical HRF regressors (one
per condition). So create a new F-contrast, call it the “Canonical HRF
effects of interest”, and enter as the weights matrix (SPM will
automatically perform zero padding over the movement regressors):
[1 0 0 0 0 0 0 0 0
0 0 0 1 0 0 0 0 0
0 0 0 0 0 0 1 0 0]
Then select “Replicate” to reproduce this across the 9 sessions. We can
also define a T-contrast to identify face-related activation, e.g.,
“Faces \(>\) Scrambled Faces”, given the weight matrix
[0.5 0 0 0.5 0 0 -1 0 0], again replicated across sessions. (Note that
there are 3 basis functions per condition, and the zeros here ignore the
temporal and dispersion derivatives, but if you want to include them,
you can add them as separate rows and test for any face-related
differences in BOLD response with an F-contrast; see earlier chapters).
Finally, for the group-level (2nd-level) analyses below, we need an estimate of activation of each condition separately (versus baseline), averaged across the sessions. So create three new T-contrasts, whose weights correspond to the three rows of the above F-contrast, i.e, that pick out the parameter estimate for the canonical HRF (with “Replicate over sessions” set to “Replicate”):
-
for Famous:
[1 0 0 0 0 0 0 0 0], -
for Unfamiliar:
[0 0 0 1 0 0 0 0 0], -
for Scrambled:
[0 0 0 0 0 0 1 0 0].
These T-contrasts will be numbered 3-5, and used in group analysis below.[^4]
Save batch and review¶
You can save this batch file (it should look like the
batch_stats_fmri_job.m file in the SPM12batch FTP directory). When
it has run, you can press “Results” from the SPM Menu window, select the
SPM.mat file in the BOLD directory, and explore some of the
contrasts. However, we will wait for the group analysis below before
showing results here.
Creating a script for analysis across subjects¶
Now that we have created a pipeline for fMRI preprocessing and analysis
for a single subject, we can script it to run on the remaining 15
subjects. Below is an example from the master_script.m:
nrun = 9;
spm_jobman('initcfg');
spm('defaults', 'FMRI');
for s = 1:nsub
%% Change to subject's directory
swd = fullfile(outpth,subdir{s},'BOLD');
cd(swd);
%% Preprocessing
jobfile = {fullfile(scrpth,'batch_preproc_fmri_job.m')};
inputs = cell(nrun+2,1);
for r = 1:nrun
inputs{r} = cellstr(spm_select('FPList',fullfile(outpth,subdir{s},'BOLD',sprintf('Run_%02d',r)),'^fMR.*\.nii$'));
end
inputs{10} = cellstr(spm_select('FPList',fullfile(outpth,subdir{s},'SMRI'),'^mprage.*\.nii$'));
inputs{11} = cellstr(spm_select('FPList',fullfile(outpth,subdir{s},'SMRI'),'^mprage.*\.nii$'));
spm_jobman('serial', jobfile, '', inputs{:});
%% 1st-level stats
jobfile = {fullfile(scrpth,'batch_stats_fmri_job.m')};
inputs = {}; %cell(nrun*3+1,1);
inputs{1} = {fullfile(swd,'Stats')};
try mkdir(inputs{1}{1}); end
for r = 1:nrun
inputs{end+1} = cellstr(spm_select('FPList',fullfile(outpth,subdir{s},'BOLD',sprintf('Run_%02d',r)),'^swfMR.*\.nii$'));
inputs{end+1} = cellstr(fullfile(outpth,subdir{s},'BOLD','Trials',sprintf('run_%02d_spmdef.mat',r)));
inputs{end+1} = cellstr(spm_select('FPList',fullfile(outpth,subdir{s},'BOLD',sprintf('Run_%02d',r)),'^rp.*\.txt$'));
end
spm_jobman('serial', jobfile, '', inputs{:});
eval(sprintf('!rm -r %s/Run*/f*',swd)) % to save diskspace (take care!)
eval(sprintf('!rm -r %s/Run*/wf*',swd)) % to save diskspace (take care!)
end
(The last two lines in the loop are optional, to delete intermediate files and save diskspace.) Once you have run this script, we can do 2nd-level (group) statistics on resulting contrast images for each condition (averaged across 9 runs).
Group Statistics on fMRI data¶
Now we have a new set of 16\(\times\)3 NIfTI images for each
subject and each condition, we can put them into the same
repeated-measures ANOVA that we used to test for differences in power
across sensors in the time-frequency analysis above, i.e, reuse the
batch_stats_rmANOVA_job.m file created above. This can be scripted as:
fmristatsdir = fullfile(outpth,'BOLD');
if ~exist(fmristatsdir)
eval(sprintf('!mkdir %s',fmristatsdir));
end
jobfile = {fullfile(scrpth,'batch_stats_rmANOVA_job.m')};
inputs = cell(nsub+1, 1);
inputs{1} = {fmristatsdir};
for s = 1:nsub
inputs{s+1,1} = cellstr(strvcat(spm_select('FPList',fullfile(outpth,subdir{s},'BOLD','Stats'),'con_000[345].nii'))); % Assumes that these T-contrasts in fMRI 1st-level models are famous, unfamiliar, scrambled (averaged across sessions)
end
spm_jobman('serial', jobfile, '', inputs{:});
Save batch and review.¶
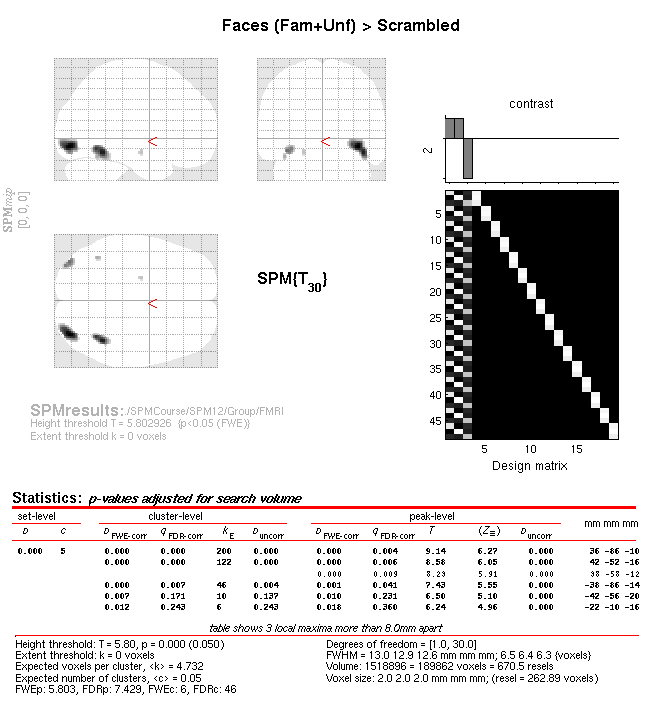
When the script has run, press “Results” from the SPM Menu window and
select the SPM.mat file in the BOLD directory. From the Contrast
Manager window, select the pre-specified T-contrast “Faces (Fam+Unf) \(>\)
Scrambled”. Within the “Stats: Results” window, when given the option,
select the following: Apply Masking: None, P value adjustment to
control: FWE, keep the threshold at 0.05, extent threshold voxels: 0;
Data Type: Volumetric 2D/3D. The Graphics window should then show what
is in Figure 1.11 below. Note the left and right
OFA and FFA (more extensive on right), plus a small cluster in left
medial temporal lobe.
Later, we can use these five clusters as priors for constraining the
inversion of our EEG/MEG data. To do this, we need to save these as an
image. Press the “save...” button in the bottom right of the SPM
Results window, and select the “all clusters (binary)” option. The
window will now prompt you for an output filename, in which you can type
in fac-scr_fmri_05_cor. This image will be output in the BOLD
directory, and we will use it later.